We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
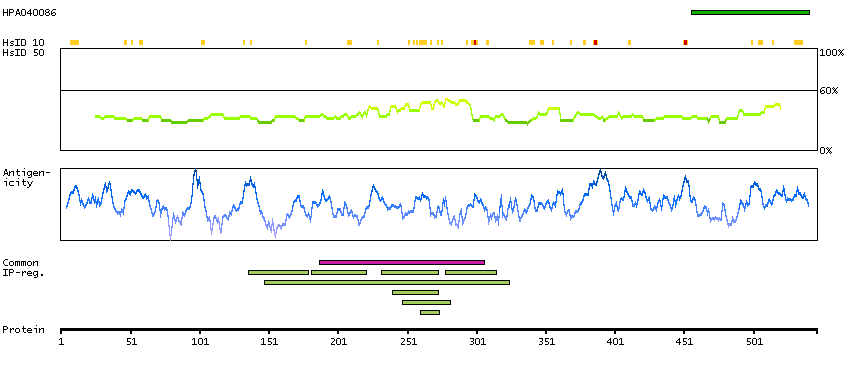
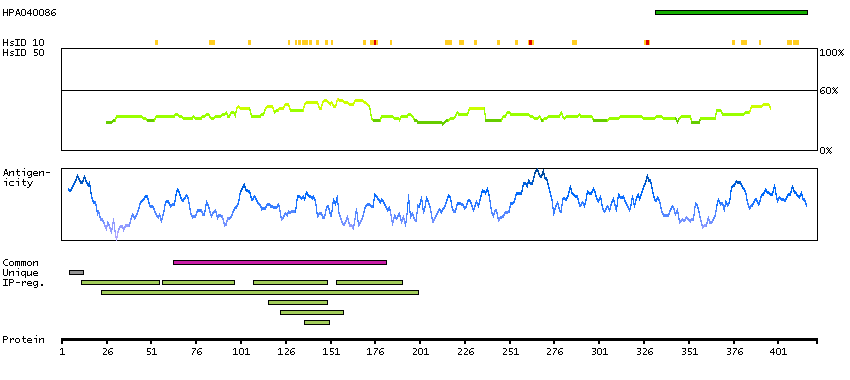
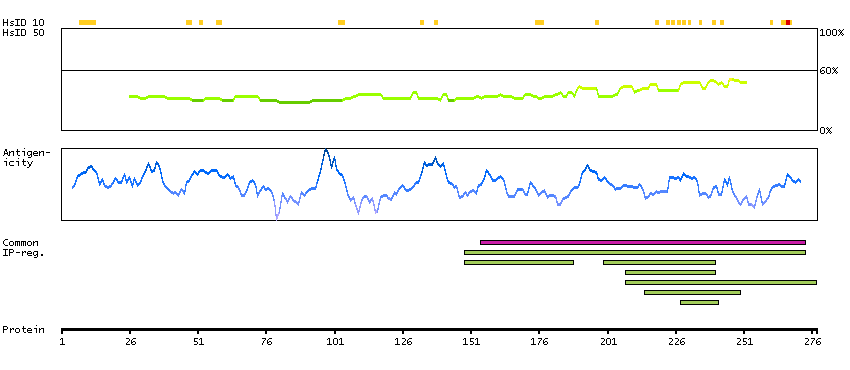
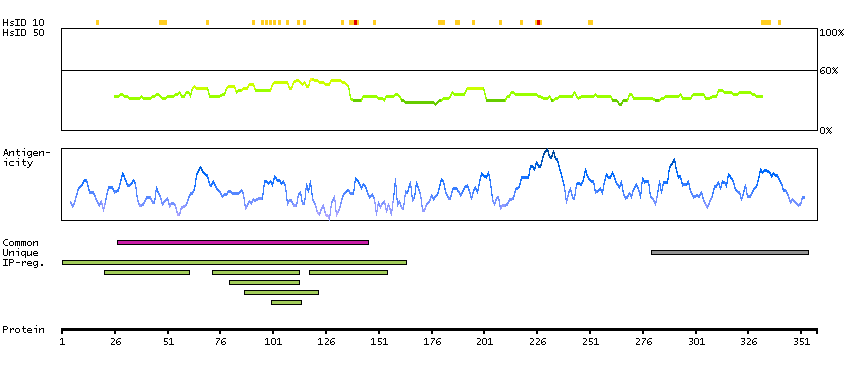
The protein encoded by this gene is a member of the WD-repeat family of regulatory proteins and may be involved in normal development of the peripheral and central nervous system. The encoded protein is part of the nuclear pore complex and is anchored there by NDC1. Defects in this gene are a cause of achalasia-addisonianism-alacrima syndrome (AAAS), also called triple-A syndrome or Allgrove syndrome. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Mar 2010]
Transporters Transporter channels and pores THUMBUP predicted membrane proteins Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Transporters Transporter channels and pores SCAMPI predicted membrane proteins THUMBUP predicted membrane proteins Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)