We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
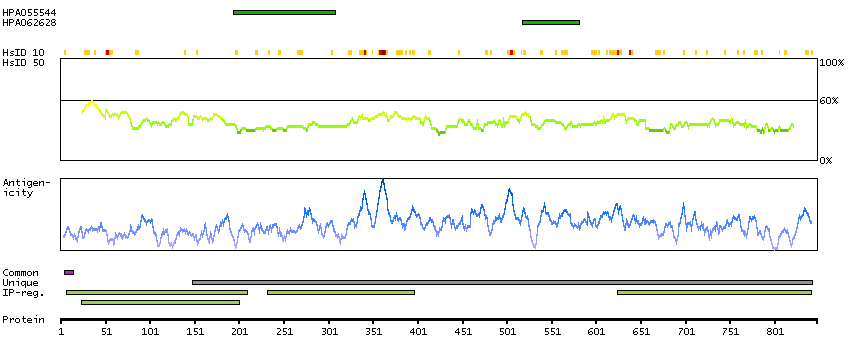
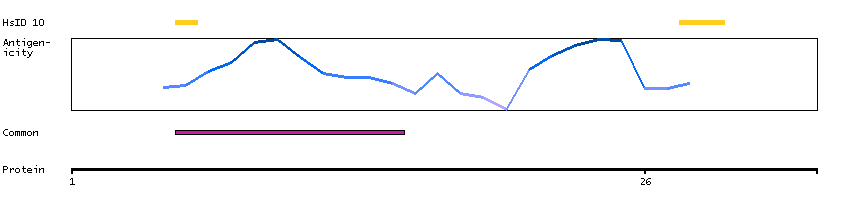
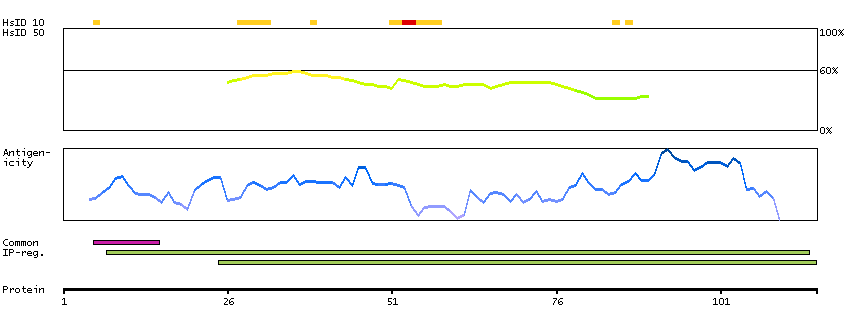
Although the function of this gene is not known, the existence of this gene is supported by mRNA and EST data. A possible function of the encoded protein can be inferred from amino acid sequence similarity to the E.coli FtsJ protein and to a mouse protein possibly involved in embryogenesis. [provided by RefSeq, Jul 2008]