We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
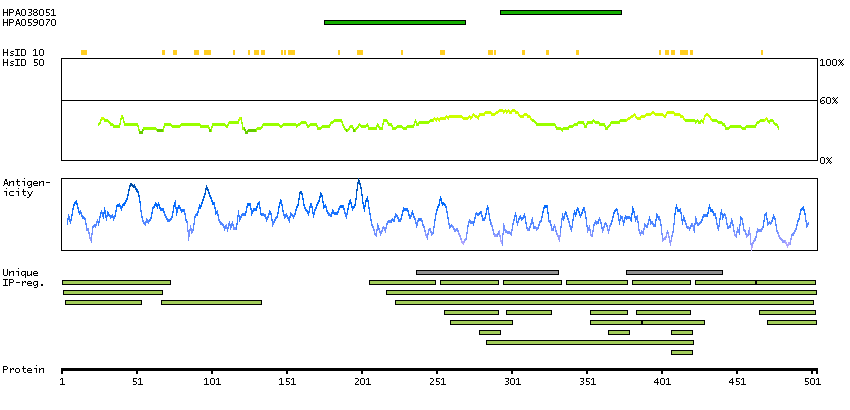
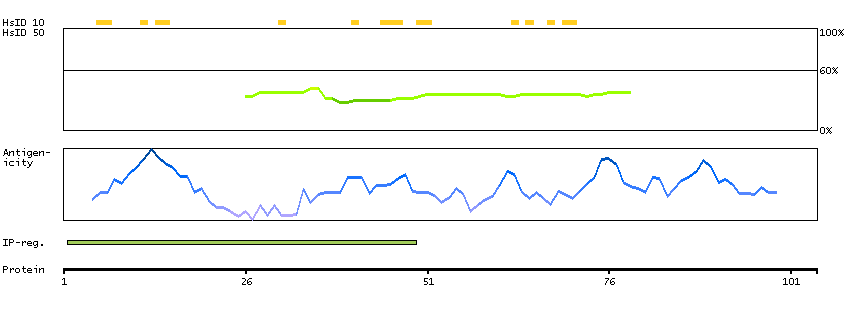
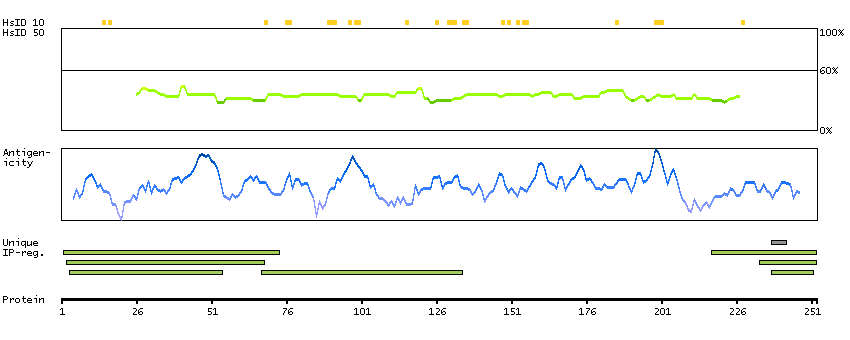
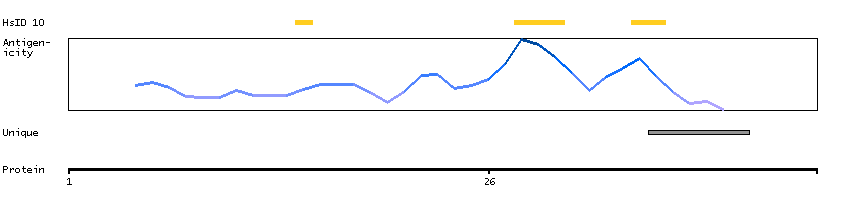
PSO4 is the human homolog of yeast Pso4, a gene essential for cell survival and DNA repair (Beck et al., 2008 [PubMed 18263876]).[supplied by OMIM, Sep 2008]