We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes an activating transcription factor, which belongs to the ATF subfamily and bZIP (basic-region leucine zipper) family. It influences cellular physiologic processes by regulating the expression of downstream target genes, which are related to growth, survival, and other cellular activities. This protein is phosphorylated at serine 63 in its kinase-inducible domain by serine/threonine kinases, cAMP-dependent protein kinase A, calmodulin-dependent protein kinase I/II, mitogen- and stress-activated protein kinase and cyclin-dependent kinase 3 (cdk-3). Its phosphorylation enhances its transactivation and transcriptional activities, and enhances cell transformation. Fusion of this gene and FUS on chromosome 16 or EWSR1 on chromosome 22 induced by translocation generates chimeric proteins in angiomatoid fibrous histiocytoma and clear cell sarcoma. This gene has a pseudogene on chromosome 6. [provided by RefSeq, Aug 2010]
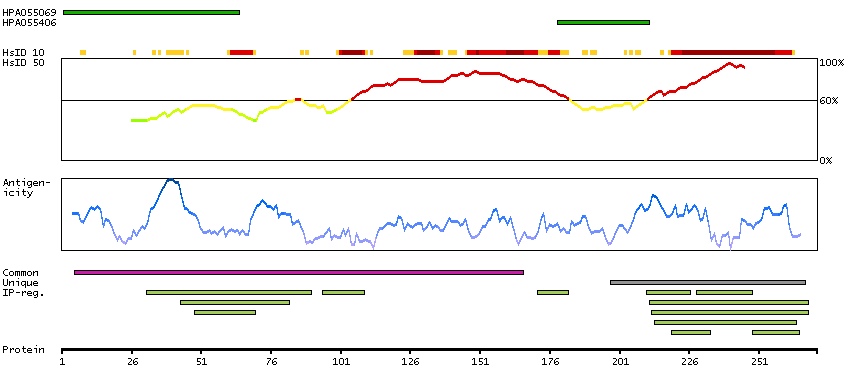
Predicted intracellular proteins Transcription factors Basic domains Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
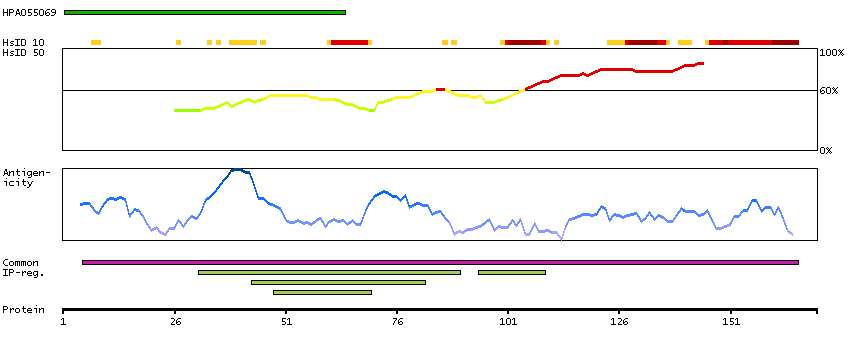
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005654 [nucleoplasm] GO:0006355 [regulation of transcription, DNA-templated]
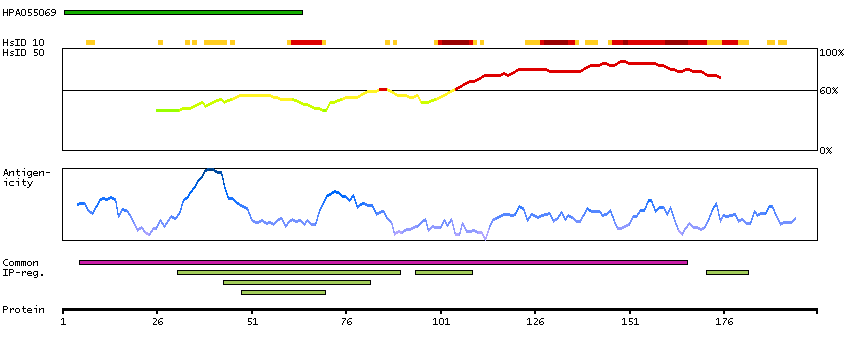
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003677 [DNA binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005654 [nucleoplasm] GO:0006355 [regulation of transcription, DNA-templated]