We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
No internal RNA expression data available for correlation. External characterization data supports antibody staining but no internal RNA data available for correlation. Paired antibodies with high similarity supports the protein profile.
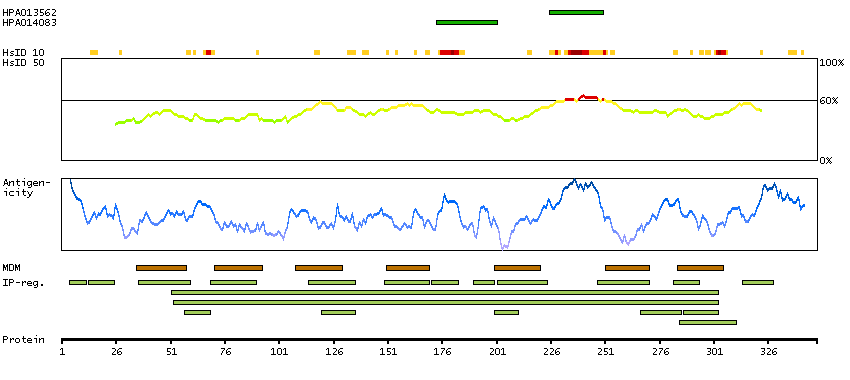
This gene belongs to the G-protein coupled receptor 1 family, opsin subfamily. It encodes the blue cone pigment gene which is one of three types of cone photoreceptors responsible for normal color vision. Defects in this gene are the cause of tritan color blindness (tritanopia). Affected individuals lack blue and yellow sensory mechanisms while retaining those for red and green. Defective blue vision is characteristic. [provided by RefSeq, Jul 2008]