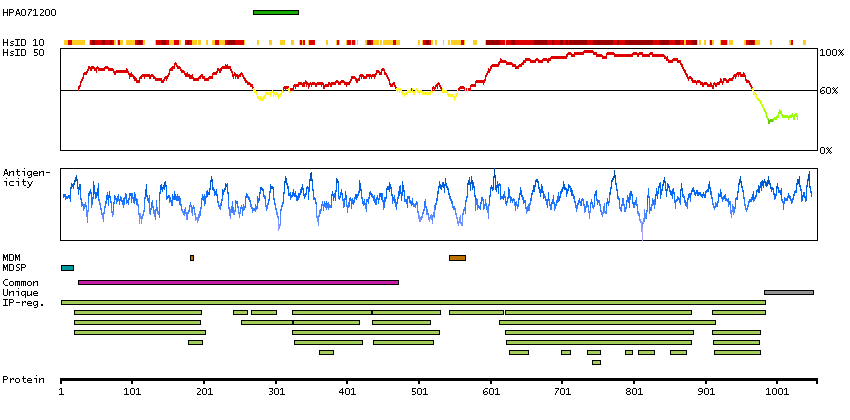
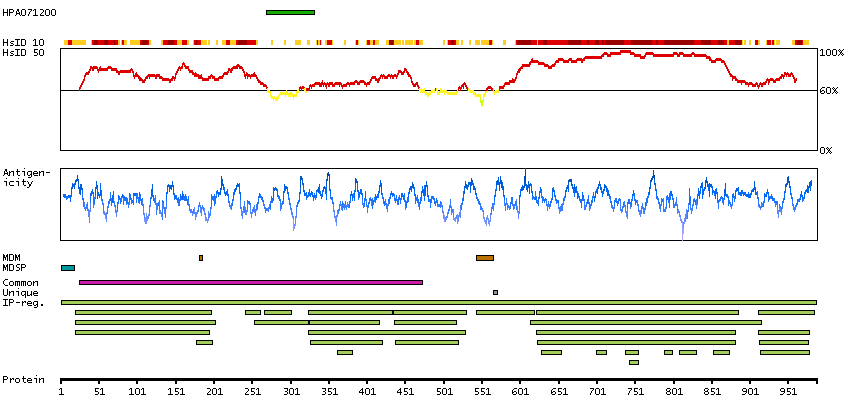
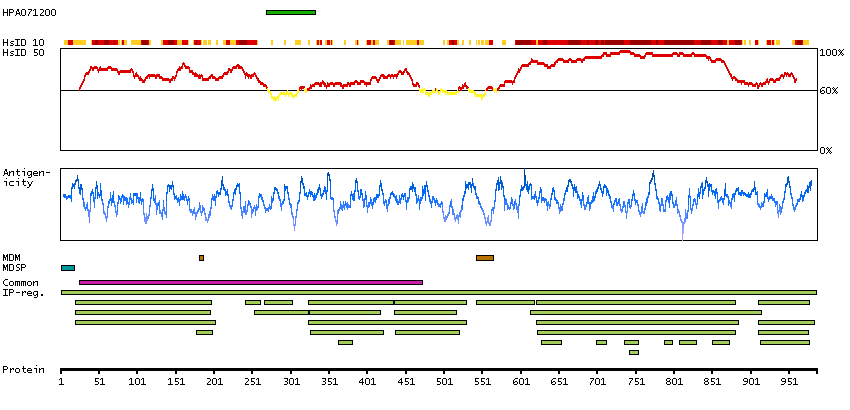
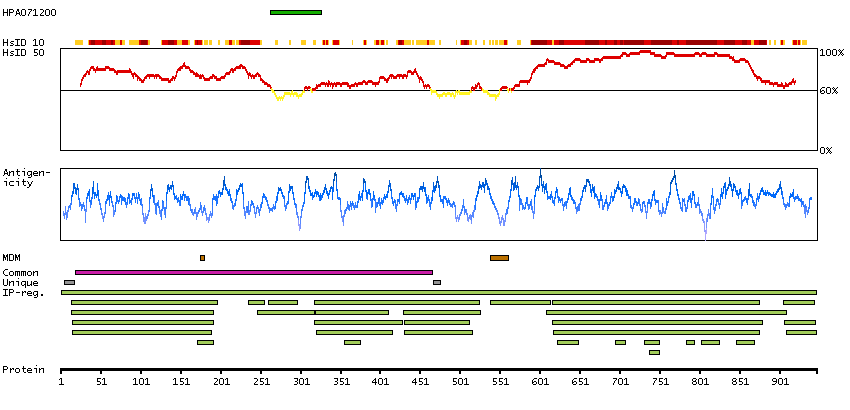
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes a member of the Eph receptor family of receptor tyrosine kinase transmembrane glycoproteins. These receptors are composed of an N-terminal glycosylated ligand-binding domain, a transmembrane region and an intracellular kinase domain. They bind ligands called ephrins and are involved in diverse cellular processes including motility, division, and differentiation. A distinguishing characteristic of Eph-ephrin signaling is that both receptors and ligands are competent to transduce a signaling cascade, resulting in bidirectional signaling. This protein belongs to a subgroup of the Eph receptors called EphB. Proteins of this subgroup are distinguished from other members of the family by sequence homology and preferential binding affinity for membrane-bound ephrin-B ligands. Allelic variants are associated with prostate and brain cancer susceptibility. Alternative splicing results in multiple transcript variants. [provided by RefSeq, May 2015]