We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Photon absorption triggers a signaling cascade in rod photoreceptors that activates cGMP phosphodiesterase (PDE), resulting in the rapid hydrolysis of cGMP, closure of cGMP-gated cation channels, and hyperpolarization of the cell. PDE is a peripheral membrane heterotrimeric enzyme made up of alpha, beta, and gamma subunits. This gene encodes the beta subunit. Mutations in this gene result in retinitis pigmentosa and autosomal dominant congenital stationary night blindness. Multiple transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Feb 2009]
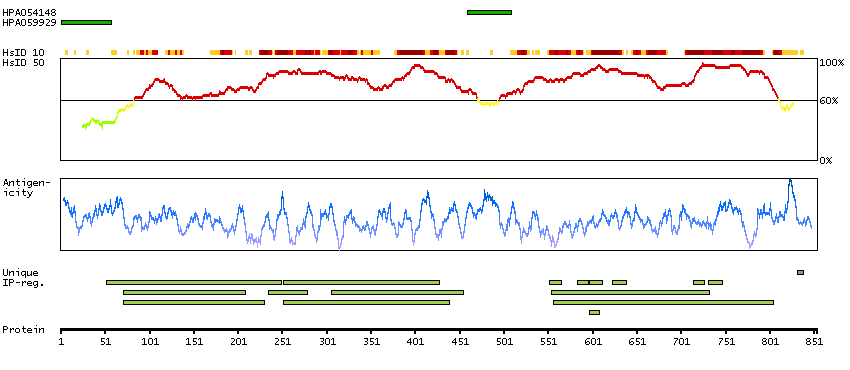
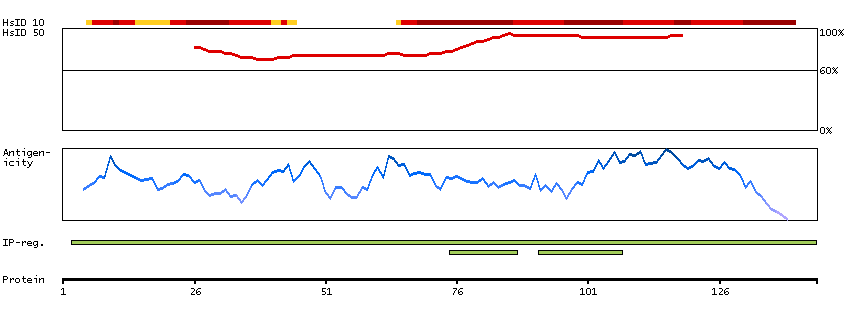
P35913 [Direct mapping] Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta
Show all
Enzymes ENZYME proteins Hydrolases Phobius predicted membrane proteins Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014)
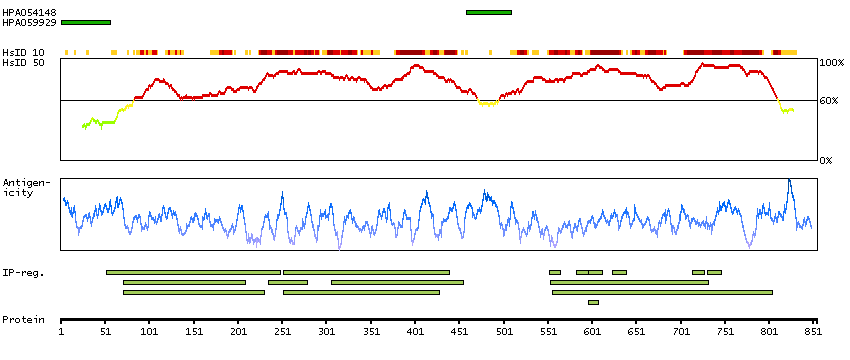
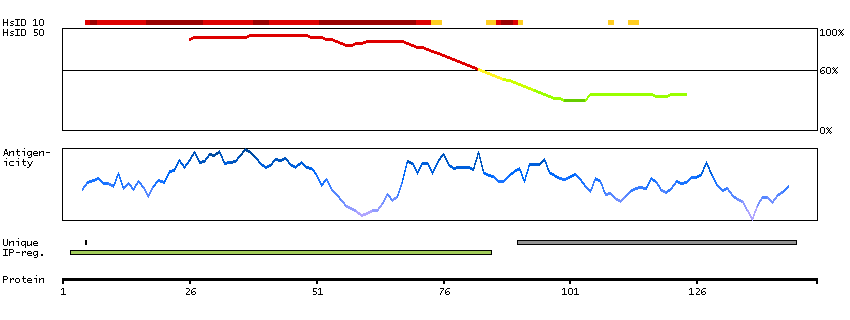
P35913 [Direct mapping] Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta
Show all
Enzymes ENZYME proteins Hydrolases Phobius predicted membrane proteins Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014)
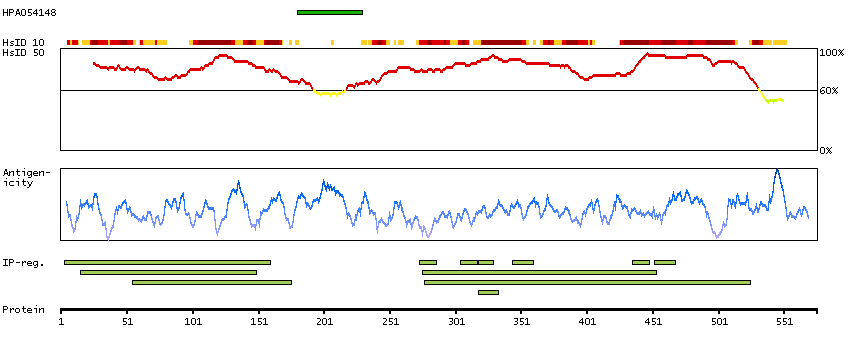
P35913 [Direct mapping] Rod cGMP-specific 3',5'-cyclic phosphodiesterase subunit beta
Show all
Enzymes ENZYME proteins Hydrolases Phobius predicted membrane proteins Predicted intracellular proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014)