We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
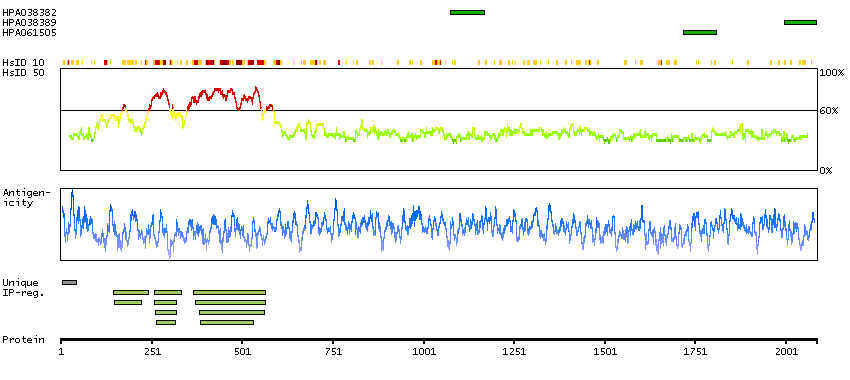
RICS is a neuron-associated GTPase-activating protein that may regulate dendritic spine morphology and strength by modulating Rho GTPase (see RHOA; MIM 165390) activity (Okabe et al., 2003 [PubMed 12531901]).[supplied by OMIM, Mar 2008]
A7KAX9 [Direct mapping] Rho GTPase-activating protein 32
Show all
Predicted intracellular proteins Cancer-related genes Mutational cancer driver genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
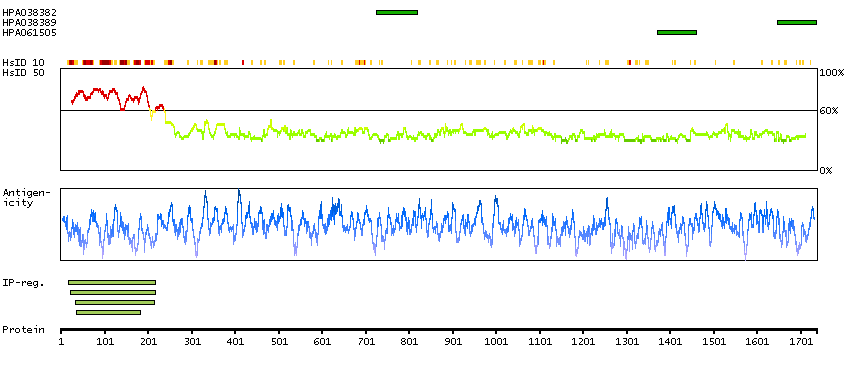
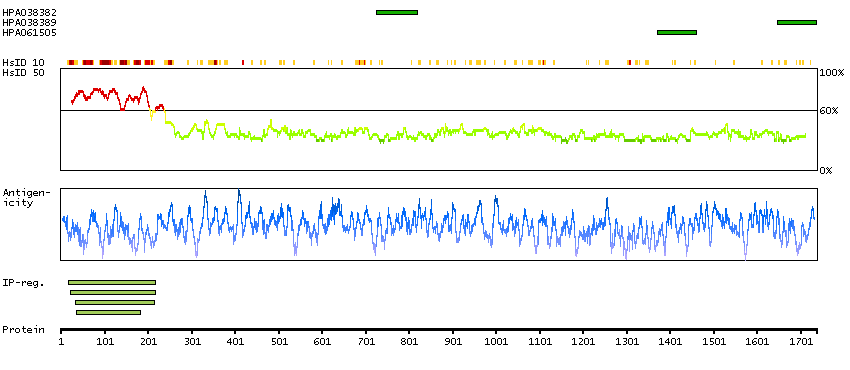
A7KAX9 [Direct mapping] Rho GTPase-activating protein 32
Show all
Predicted intracellular proteins Cancer-related genes Mutational cancer driver genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)