We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
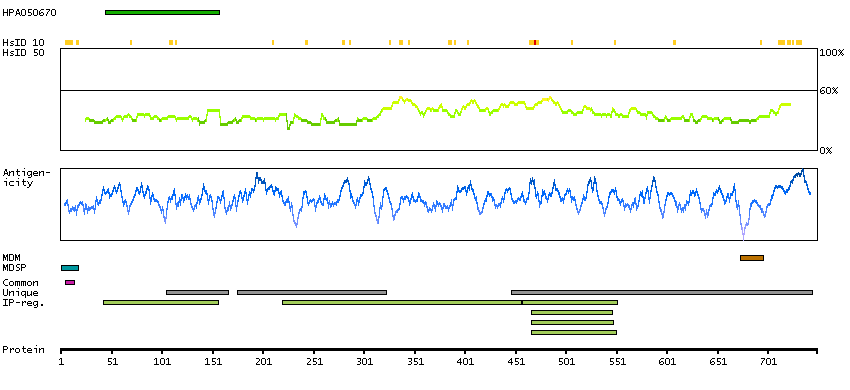
Members of the ADAM family are cell surface proteins with a unique structure possessing both potential adhesion and protease domains. This gene encodes and ADAM family member that cleaves many proteins including TNF-alpha and E-cadherin. [provided by RefSeq, Jul 2008]