We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
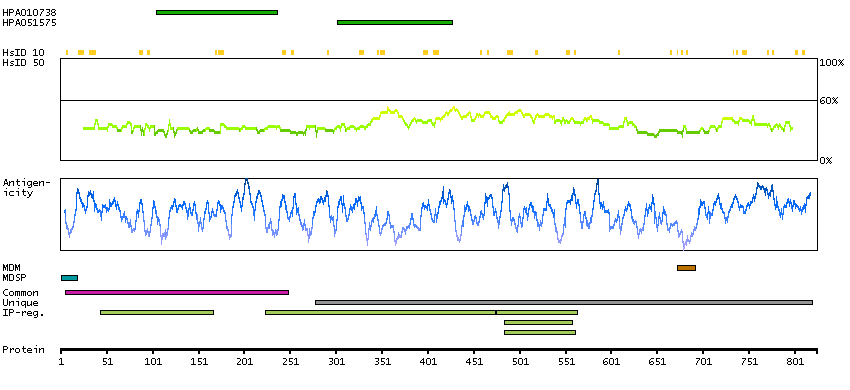
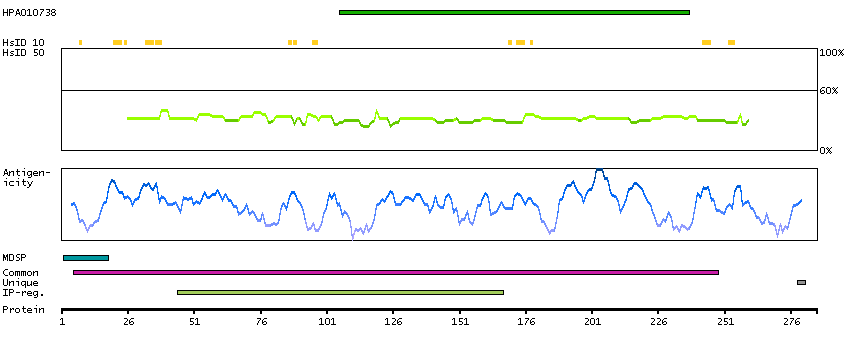
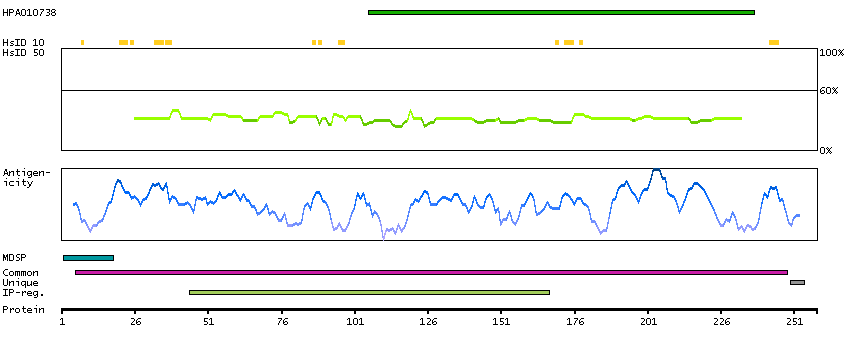
This gene encodes a member of the ADAM (a disintegrin and metalloprotease domain) family. Members of this family are membrane-anchored proteins structurally related to snake venom disintegrins, and have been implicated in a variety of biologic processes involving cell-cell and cell-matrix interactions, including fertilization, muscle development, and neurogenesis. The protein encoded by this gene functions as a tumor necrosis factor-alpha converting enzyme; binds mitotic arrest deficient 2 protein; and also plays a prominent role in the activation of the Notch signaling pathway. [provided by RefSeq, Jul 2008]