We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Polymerase (RNA) II (DNA directed) polypeptide H (HGNC Symbol)
Entrez gene summary
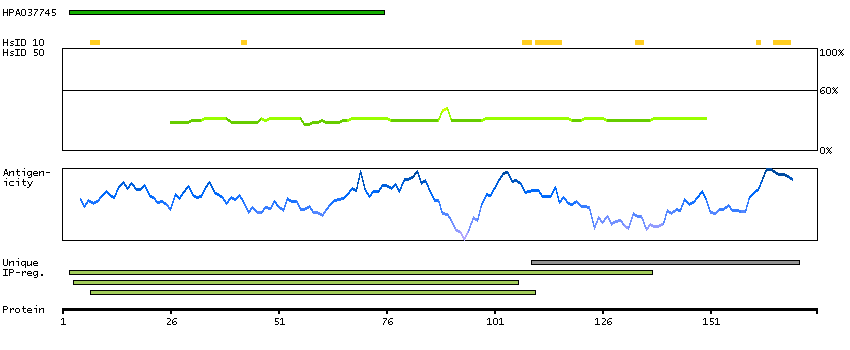
The three eukaryotic RNA polymerases are complex multisubunit enzymes that play a central role in the transcription of nuclear genes. This gene encodes an essential and highly conserved subunit of RNA polymerase II that is shared by the other two eukaryotic DNA-directed RNA polymerases, I and III. Alternative splicing results in multiple transcript variants of this gene. [provided by RefSeq, Jul 2013]
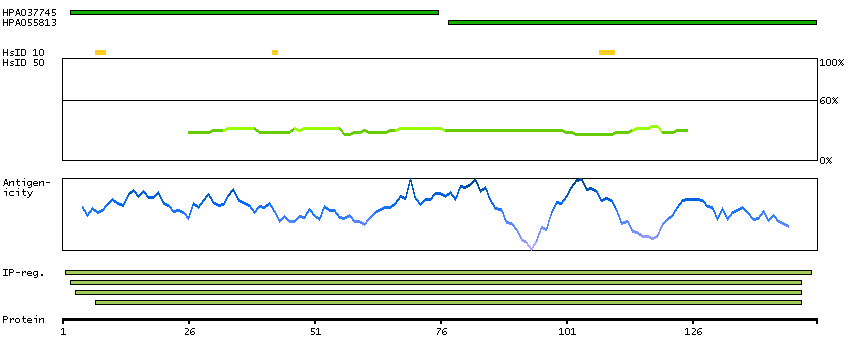
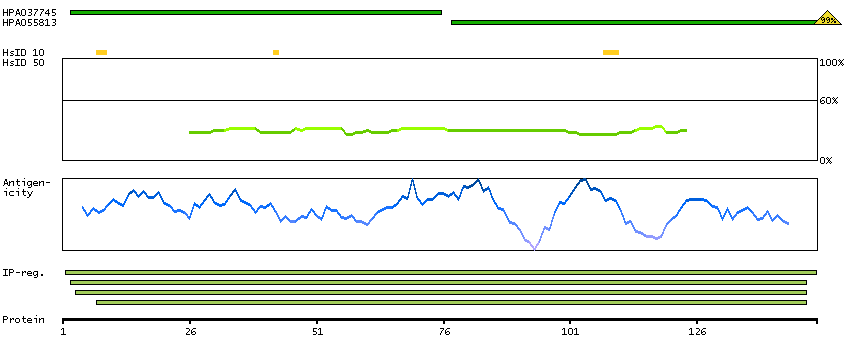
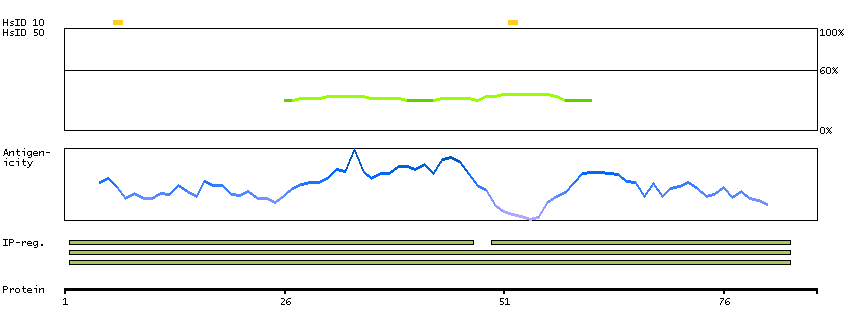
C9JJJ9 [Direct mapping] DNA-directed RNA polymerases I, II, and III subunit RPABC3
Show all
Predicted intracellular proteins RNA polymerase related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001054 [RNA polymerase I activity] GO:0001055 [RNA polymerase II activity] GO:0001056 [RNA polymerase III activity] GO:0005666 [DNA-directed RNA polymerase III complex] GO:0005736 [DNA-directed RNA polymerase I complex] GO:0006360 [transcription from RNA polymerase I promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006383 [transcription from RNA polymerase III promoter]
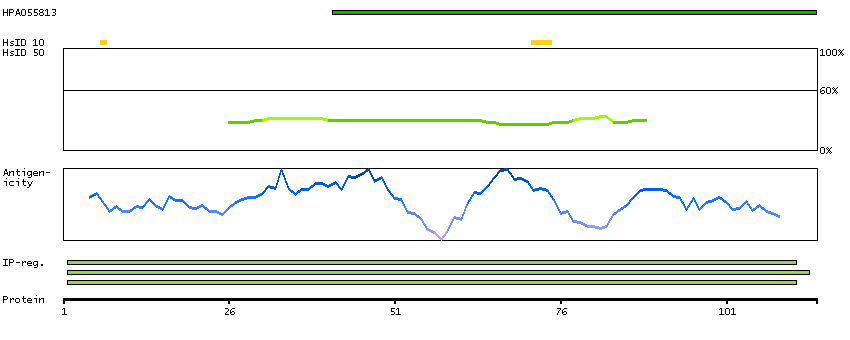
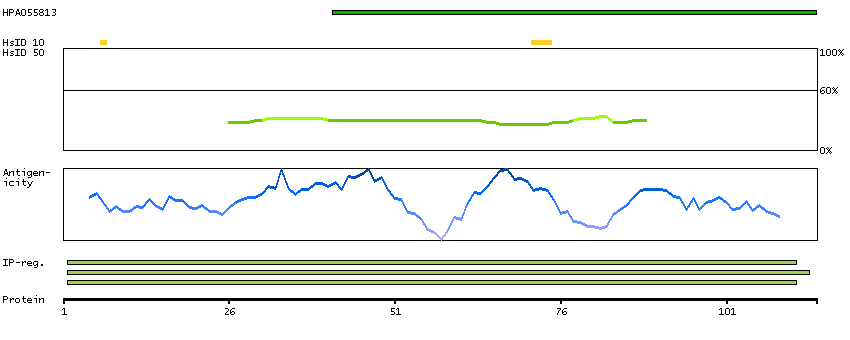
C9JLU1 [Direct mapping] DNA-directed RNA polymerases I, II, and III subunit RPABC3
Show all
Predicted intracellular proteins RNA polymerase related proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0001054 [RNA polymerase I activity] GO:0001055 [RNA polymerase II activity] GO:0001056 [RNA polymerase III activity] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005666 [DNA-directed RNA polymerase III complex] GO:0005736 [DNA-directed RNA polymerase I complex] GO:0006351 [transcription, DNA-templated] GO:0006360 [transcription from RNA polymerase I promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0006383 [transcription from RNA polymerase III promoter]