We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
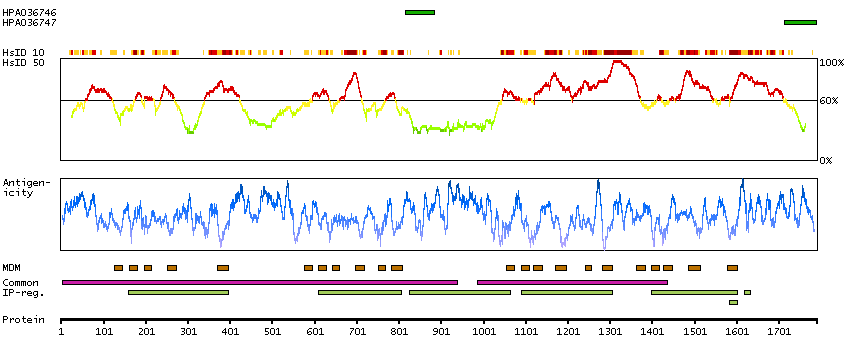
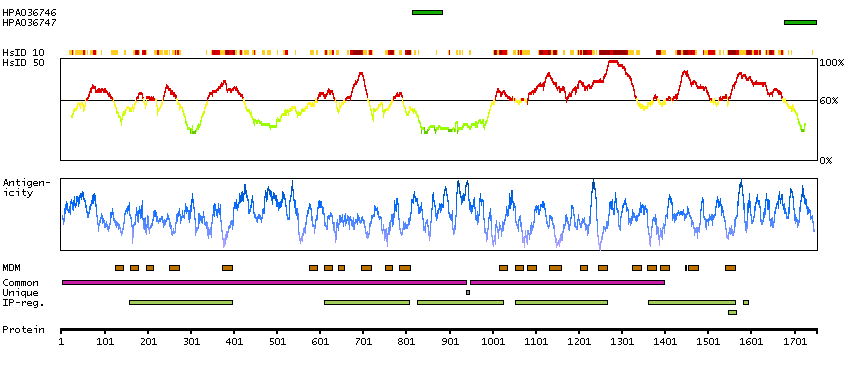
Sodium channel, voltage gated, type XI alpha subunit (HGNC Symbol)
Entrez gene summary
Voltage-gated sodium channels are membrane protein complexes that play a fundamental role in the rising phase of the action potential in most excitable cells. Alpha subunits, such as SCN11A, mediate voltage-dependent gating and conductance, while auxiliary beta subunits regulate the kinetic properties of the channel and facilitate membrane localization of the complex. Aberrant expression patterns or mutations of alpha subunits underlie a number of disorders. Each alpha subunit consists of 4 domains connected by 3 intracellular loops; each domain consists of 6 transmembrane segments and intra- and extracellular linkers.[supplied by OMIM, Apr 2004]