We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
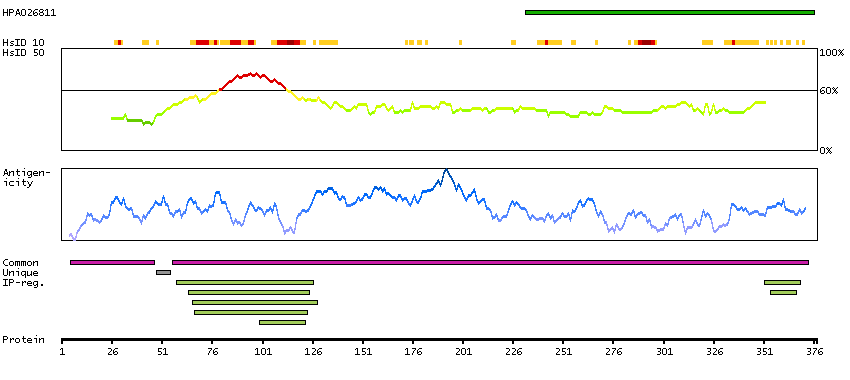
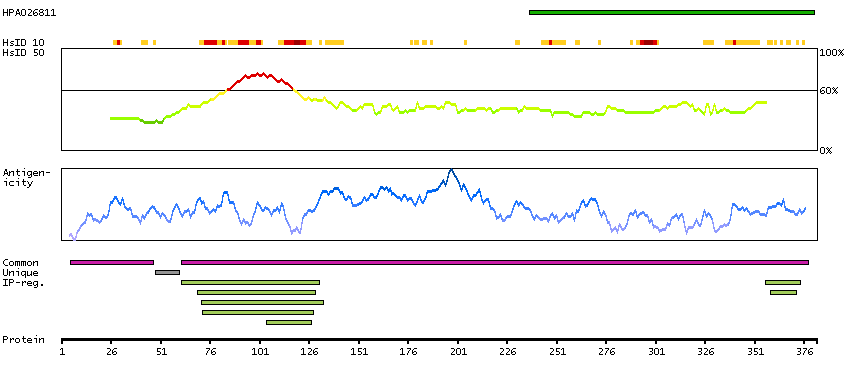
This gene encodes a member of the bicoid sub-family of homeodomain-containing transcription factors. The encoded protein acts as a transcription factor and may play a role in brain and sensory organ development. Two transcript variants encoding distinct isoforms have been identified for this gene. [provided by RefSeq, Jul 2008]