We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
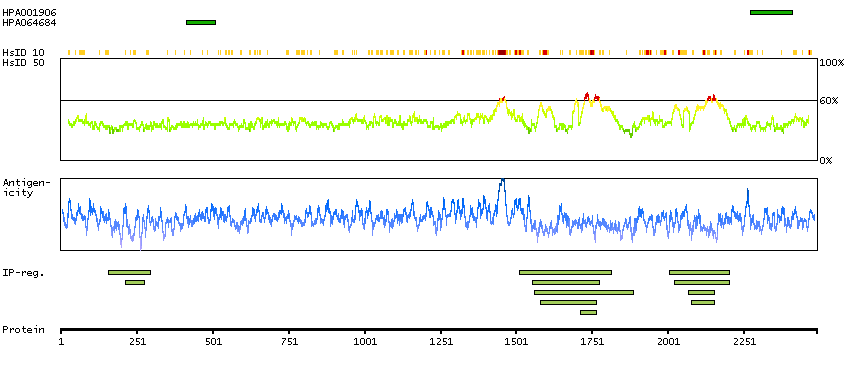
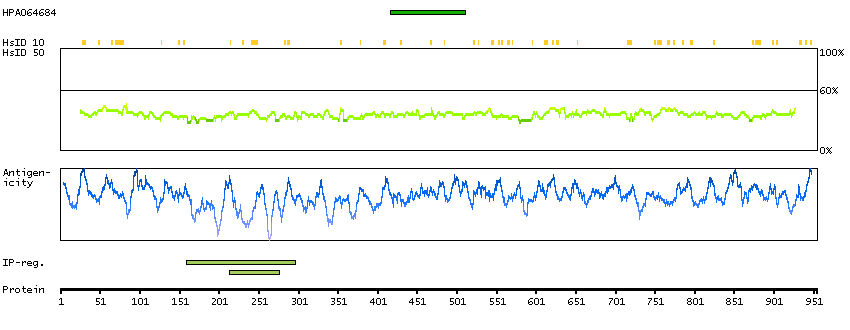
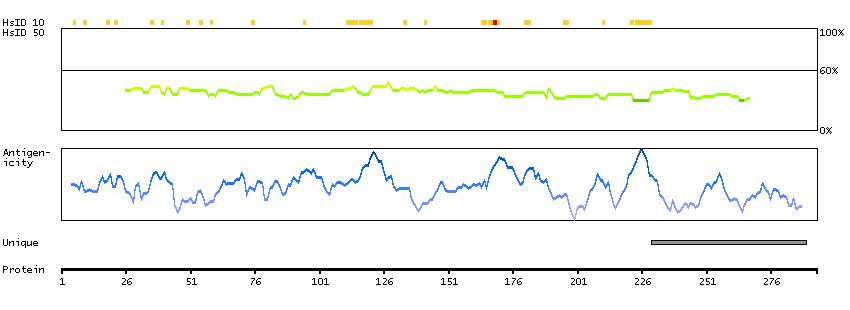
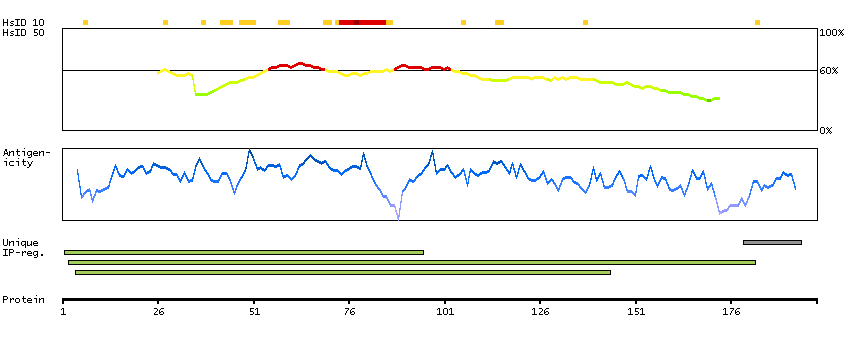
The protein encoded by this gene contains an ATPase/helicase domain, and thus it belongs to the SWI/SNF family of chromatin remodeling proteins. This protein is found to undergo cell cycle-dependent phosphorylation, which regulates its nuclear matrix and chromatin association, and suggests its involvement in the gene regulation at interphase and chromosomal segregation in mitosis. Mutations in this gene are associated with an X-linked mental retardation (XLMR) syndrome most often accompanied by alpha-thalassemia (ATRX) syndrome. These mutations have been shown to cause diverse changes in the pattern of DNA methylation, which may provide a link between chromatin remodeling, DNA methylation, and gene expression in developmental processes. Multiple alternatively spliced transcript variants encoding distinct isoforms have been reported. [provided by RefSeq, Aug 2013]