We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
HMG-CoA reductase is the rate-limiting enzyme for cholesterol synthesis and is regulated via a negative feedback mechanism mediated by sterols and non-sterol metabolites derived from mevalonate, the product of the reaction catalyzed by reductase. Normally in mammalian cells this enzyme is suppressed by cholesterol derived from the internalization and degradation of low density lipoprotein (LDL) via the LDL receptor. Competitive inhibitors of the reductase induce the expression of LDL receptors in the liver, which in turn increases the catabolism of plasma LDL and lowers the plasma concentration of cholesterol, an important determinant of atherosclerosis. Alternatively spliced transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Aug 2008]
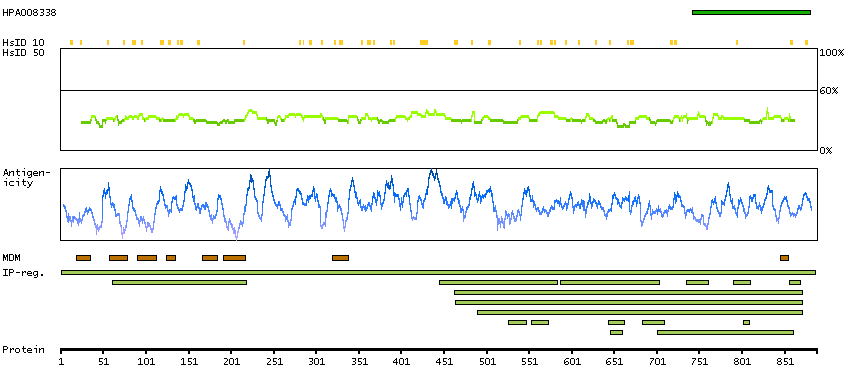
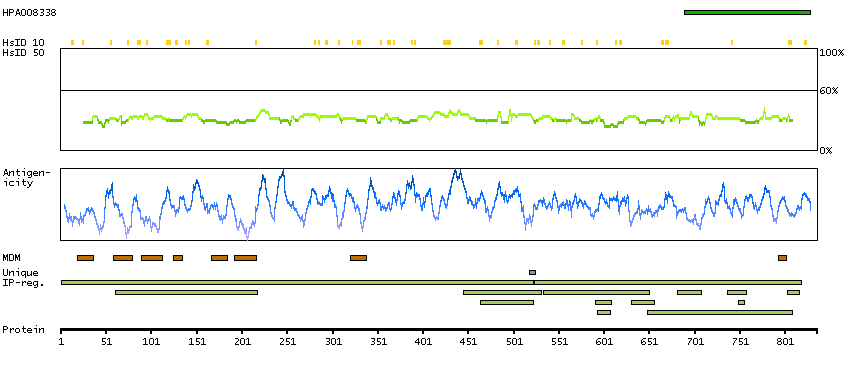
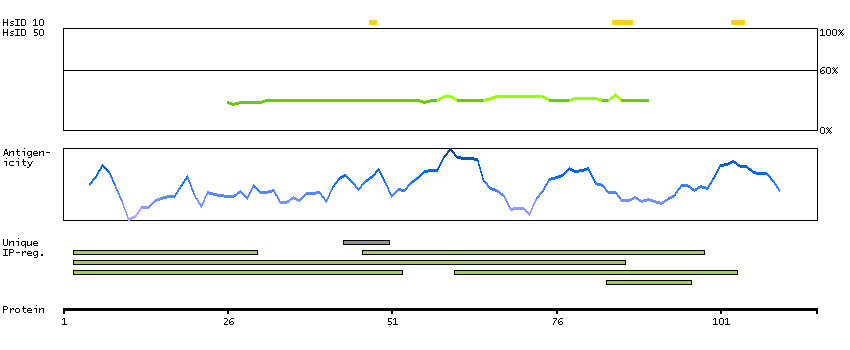
H0Y8K6 [Direct mapping] 3-hydroxy-3-methylglutaryl-coenzyme A reductase
Show all
SCAMPI predicted membrane proteins SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0004420 [hydroxymethylglutaryl-CoA reductase (NADPH) activity] GO:0005622 [intracellular] GO:0008299 [isoprenoid biosynthetic process] GO:0015936 [coenzyme A metabolic process] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0050662 [coenzyme binding] GO:0055114 [oxidation-reduction process]
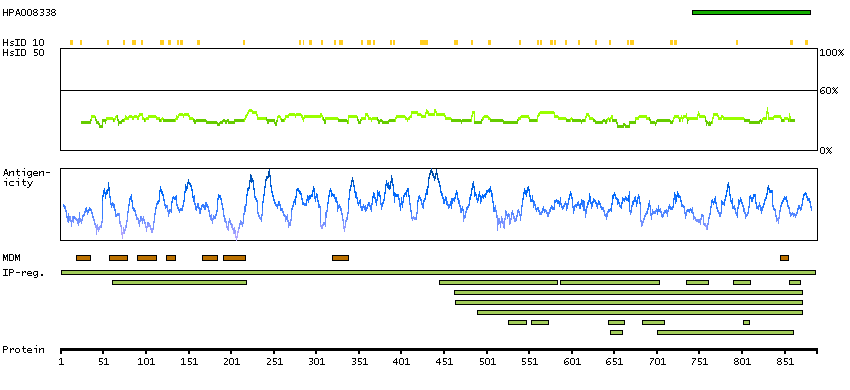
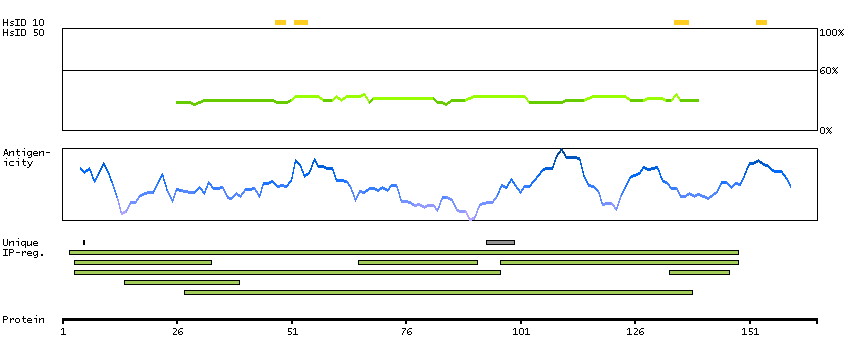
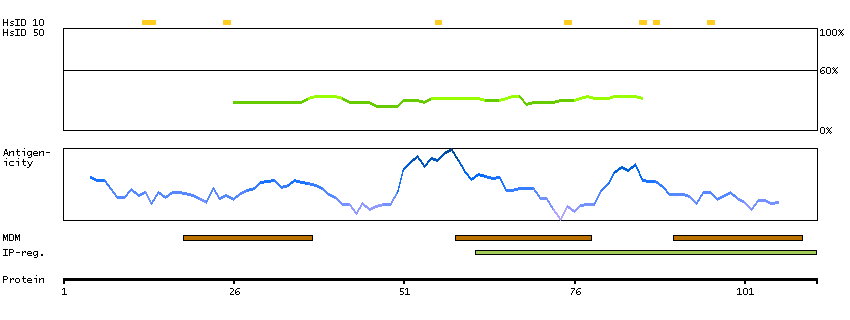
H0Y8F6 [Direct mapping] 3-hydroxy-3-methylglutaryl-coenzyme A reductase
Show all
Phobius predicted membrane proteins SPOCTOPUS predicted membrane proteins Predicted intracellular proteins Protein evidence (Ezkurdia et al 2014)
Show all
GO:0004420 [hydroxymethylglutaryl-CoA reductase (NADPH) activity] GO:0005622 [intracellular] GO:0008299 [isoprenoid biosynthetic process] GO:0015936 [coenzyme A metabolic process] GO:0016616 [oxidoreductase activity, acting on the CH-OH group of donors, NAD or NADP as acceptor] GO:0050662 [coenzyme binding] GO:0055114 [oxidation-reduction process]