We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
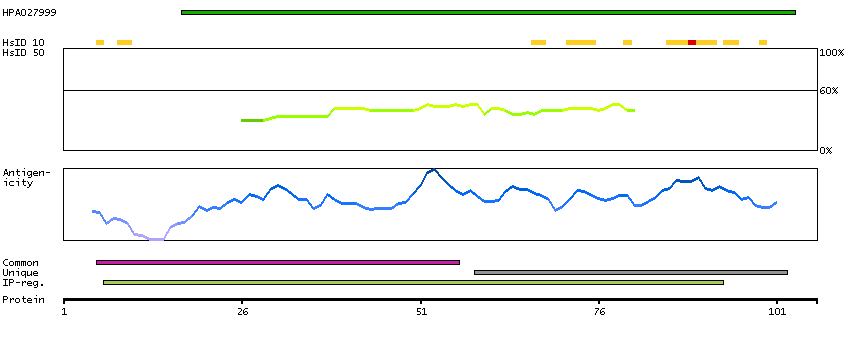
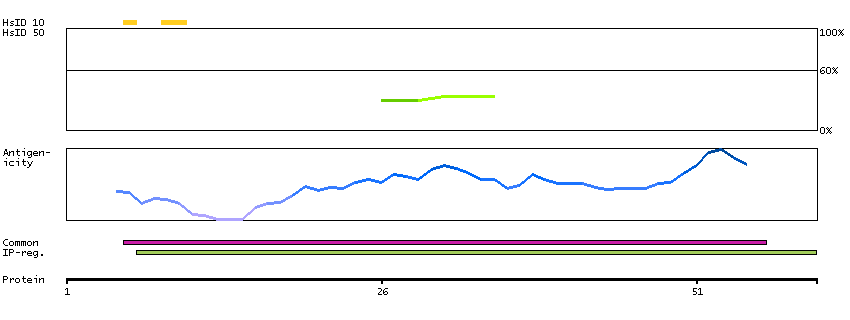
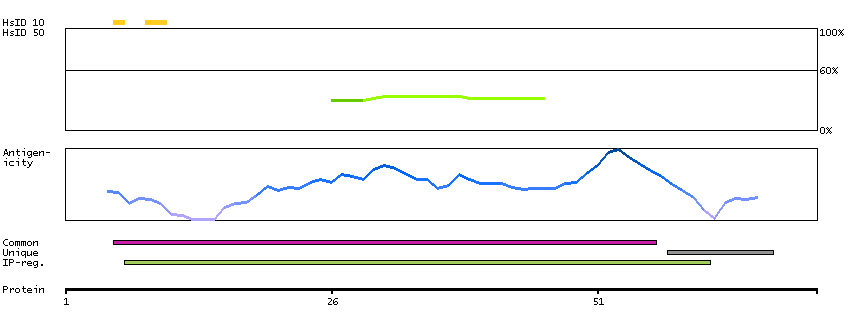
This gene encodes a mitochondrial ATPase inhibitor. Alternative splicing occurs at this locus and three transcript variants encoding distinct isoforms have been identified. [provided by RefSeq, Jul 2008]