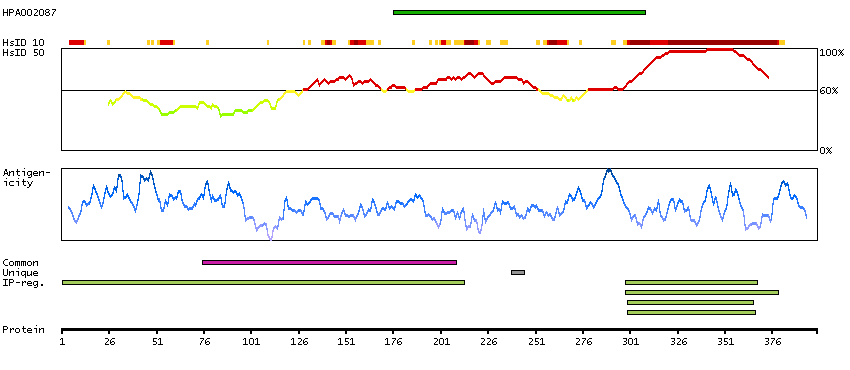
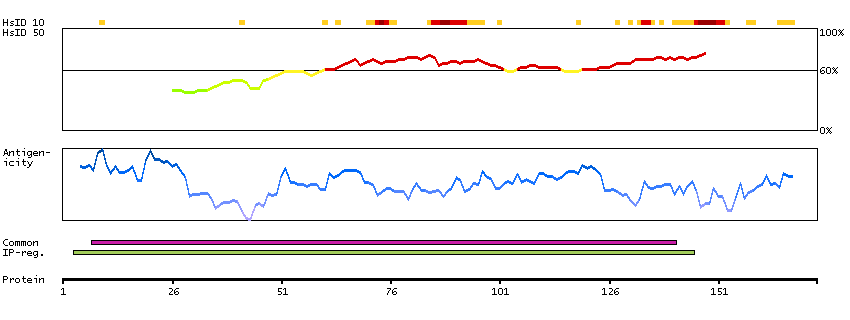
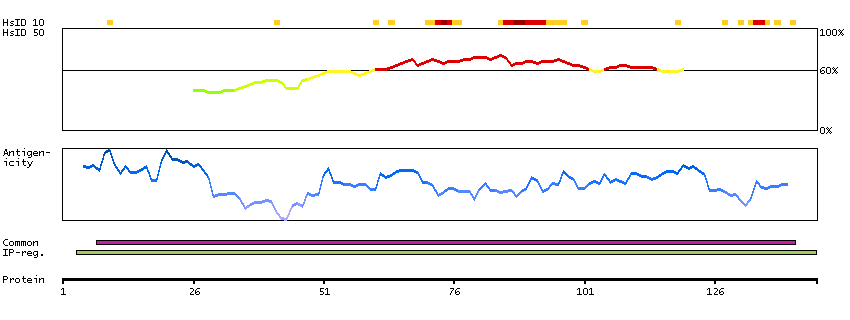
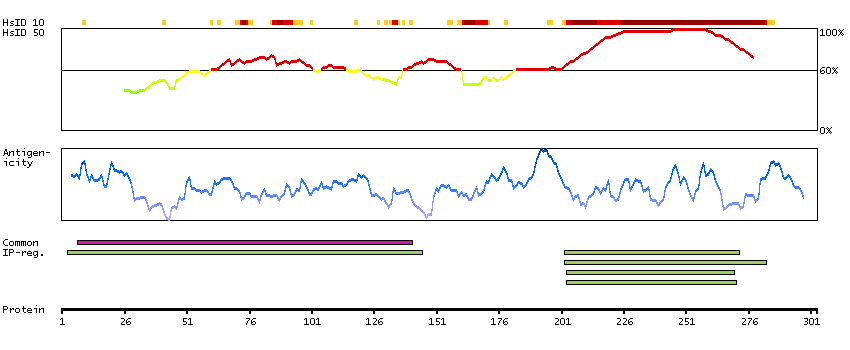
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
This gene encodes a transcription factor belonging to a family of proteins that share homology with the high mobility group protein-1. The protein encoded by this gene can bind to a functionally important site in the T-cell receptor-alpha enhancer, thereby conferring maximal enhancer activity. This transcription factor is involved in the Wnt signaling pathway, and it may function in hair cell differentiation and follicle morphogenesis. Mutations in this gene have been found in somatic sebaceous tumors. This gene has also been linked to other cancers, including androgen-independent prostate cancer. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Oct 2009]
Predicted intracellular proteins Plasma proteins Transcription factors Other all-alpha-helical DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000122 [negative regulation of transcription from RNA polymerase II promoter] GO:0000977 [RNA polymerase II regulatory region sequence-specific DNA binding] GO:0000978 [RNA polymerase II core promoter proximal region sequence-specific DNA binding] GO:0001077 [transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding] GO:0001228 [transcriptional activator activity, RNA polymerase II transcription regulatory region sequence-specific binding] GO:0001569 [patterning of blood vessels] GO:0001649 [osteoblast differentiation] GO:0001755 [neural crest cell migration] GO:0001756 [somitogenesis] GO:0001822 [kidney development] GO:0001837 [epithelial to mesenchymal transition] GO:0001944 [vasculature development] GO:0002040 [sprouting angiogenesis] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003705 [transcription factor activity, RNA polymerase II distal enhancer sequence-specific binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005667 [transcription factor complex] GO:0005737 [cytoplasm] GO:0006357 [regulation of transcription from RNA polymerase II promoter] GO:0006366 [transcription from RNA polymerase II promoter] GO:0007155 [cell adhesion] GO:0008013 [beta-catenin binding] GO:0008134 [transcription factor binding] GO:0008284 [positive regulation of cell proliferation] GO:0008301 [DNA binding, bending] GO:0010226 [response to lithium ion] GO:0010628 [positive regulation of gene expression] GO:0010718 [positive regulation of epithelial to mesenchymal transition] GO:0014070 [response to organic cyclic compound] GO:0016055 [Wnt signaling pathway] GO:0016202 [regulation of striated muscle tissue development] GO:0021542 [dentate gyrus development] GO:0021766 [hippocampus development] GO:0021854 [hypothalamus development] GO:0021861 [forebrain radial glial cell differentiation] GO:0021873 [forebrain neuroblast division] GO:0021879 [forebrain neuron differentiation] GO:0021943 [formation of radial glial scaffolds] GO:0022407 [regulation of cell-cell adhesion] GO:0022408 [negative regulation of cell-cell adhesion] GO:0022409 [positive regulation of cell-cell adhesion] GO:0030111 [regulation of Wnt signaling pathway] GO:0030223 [neutrophil differentiation] GO:0030284 [estrogen receptor activity] GO:0030307 [positive regulation of cell growth] GO:0030326 [embryonic limb morphogenesis] GO:0030331 [estrogen receptor binding] GO:0030335 [positive regulation of cell migration] GO:0030509 [BMP signaling pathway] GO:0030854 [positive regulation of granulocyte differentiation] GO:0030879 [mammary gland development] GO:0031100 [organ regeneration] GO:0032696 [negative regulation of interleukin-13 production] GO:0032713 [negative regulation of interleukin-4 production] GO:0032714 [negative regulation of interleukin-5 production] GO:0032993 [protein-DNA complex] GO:0033153 [T cell receptor V(D)J recombination] GO:0035326 [enhancer binding] GO:0042100 [B cell proliferation] GO:0042393 [histone binding] GO:0042475 [odontogenesis of dentin-containing tooth] GO:0043027 [cysteine-type endopeptidase inhibitor activity involved in apoptotic process] GO:0043066 [negative regulation of apoptotic process] GO:0043154 [negative regulation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0043392 [negative regulation of DNA binding] GO:0043401 [steroid hormone mediated signaling pathway] GO:0043565 [sequence-specific DNA binding] GO:0043586 [tongue development] GO:0043588 [skin development] GO:0043923 [positive regulation by host of viral transcription] GO:0043966 [histone H3 acetylation] GO:0043967 [histone H4 acetylation] GO:0044212 [transcription regulatory region DNA binding] GO:0045063 [T-helper 1 cell differentiation] GO:0045295 [gamma-catenin binding] GO:0045843 [negative regulation of striated muscle tissue development] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046632 [alpha-beta T cell differentiation] GO:0048069 [eye pigmentation] GO:0048341 [paraxial mesoderm formation] GO:0048468 [cell development] GO:0048699 [generation of neurons] GO:0048747 [muscle fiber development] GO:0050909 [sensory perception of taste] GO:0060021 [palate development] GO:0060033 [anatomical structure regression] GO:0060070 [canonical Wnt signaling pathway] GO:0060325 [face morphogenesis] GO:0060326 [cell chemotaxis] GO:0060561 [apoptotic process involved in morphogenesis] GO:0060710 [chorio-allantoic fusion] GO:0061153 [trachea gland development] GO:0070016 [armadillo repeat domain binding] GO:0070742 [C2H2 zinc finger domain binding] GO:0071345 [cellular response to cytokine stimulus] GO:0071353 [cellular response to interleukin-4] GO:0071864 [positive regulation of cell proliferation in bone marrow] GO:0071866 [negative regulation of apoptotic process in bone marrow] GO:0071895 [odontoblast differentiation] GO:0071899 [negative regulation of estrogen receptor binding] GO:0090068 [positive regulation of cell cycle process] GO:0090090 [negative regulation of canonical Wnt signaling pathway] GO:1902262 [apoptotic process involved in patterning of blood vessels]
Predicted intracellular proteins Plasma proteins Transcription factors Other all-alpha-helical DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000977 [RNA polymerase II regulatory region sequence-specific DNA binding] GO:0001228 [transcriptional activator activity, RNA polymerase II transcription regulatory region sequence-specific binding] GO:0001569 [patterning of blood vessels] GO:0001649 [osteoblast differentiation] GO:0001755 [neural crest cell migration] GO:0001756 [somitogenesis] GO:0001837 [epithelial to mesenchymal transition] GO:0002040 [sprouting angiogenesis] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003705 [transcription factor activity, RNA polymerase II distal enhancer sequence-specific binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005667 [transcription factor complex] GO:0005737 [cytoplasm] GO:0006366 [transcription from RNA polymerase II promoter] GO:0008013 [beta-catenin binding] GO:0008284 [positive regulation of cell proliferation] GO:0008301 [DNA binding, bending] GO:0010628 [positive regulation of gene expression] GO:0010718 [positive regulation of epithelial to mesenchymal transition] GO:0016055 [Wnt signaling pathway] GO:0016202 [regulation of striated muscle tissue development] GO:0021542 [dentate gyrus development] GO:0021854 [hypothalamus development] GO:0021861 [forebrain radial glial cell differentiation] GO:0021873 [forebrain neuroblast division] GO:0021879 [forebrain neuron differentiation] GO:0021943 [formation of radial glial scaffolds] GO:0022408 [negative regulation of cell-cell adhesion] GO:0022409 [positive regulation of cell-cell adhesion] GO:0030223 [neutrophil differentiation] GO:0030284 [estrogen receptor activity] GO:0030307 [positive regulation of cell growth] GO:0030326 [embryonic limb morphogenesis] GO:0030331 [estrogen receptor binding] GO:0030335 [positive regulation of cell migration] GO:0030509 [BMP signaling pathway] GO:0030854 [positive regulation of granulocyte differentiation] GO:0030879 [mammary gland development] GO:0032696 [negative regulation of interleukin-13 production] GO:0032713 [negative regulation of interleukin-4 production] GO:0032714 [negative regulation of interleukin-5 production] GO:0032993 [protein-DNA complex] GO:0035326 [enhancer binding] GO:0042100 [B cell proliferation] GO:0042393 [histone binding] GO:0043027 [cysteine-type endopeptidase inhibitor activity involved in apoptotic process] GO:0043066 [negative regulation of apoptotic process] GO:0043154 [negative regulation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0043392 [negative regulation of DNA binding] GO:0043401 [steroid hormone mediated signaling pathway] GO:0043565 [sequence-specific DNA binding] GO:0043923 [positive regulation by host of viral transcription] GO:0043966 [histone H3 acetylation] GO:0043967 [histone H4 acetylation] GO:0044212 [transcription regulatory region DNA binding] GO:0045063 [T-helper 1 cell differentiation] GO:0045295 [gamma-catenin binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046632 [alpha-beta T cell differentiation] GO:0048069 [eye pigmentation] GO:0048341 [paraxial mesoderm formation] GO:0048699 [generation of neurons] GO:0048747 [muscle fiber development] GO:0060021 [palate development] GO:0060070 [canonical Wnt signaling pathway] GO:0060325 [face morphogenesis] GO:0060326 [cell chemotaxis] GO:0060561 [apoptotic process involved in morphogenesis] GO:0060710 [chorio-allantoic fusion] GO:0061153 [trachea gland development] GO:0070016 [armadillo repeat domain binding] GO:0070742 [C2H2 zinc finger domain binding] GO:0071345 [cellular response to cytokine stimulus] GO:0071353 [cellular response to interleukin-4] GO:0071864 [positive regulation of cell proliferation in bone marrow] GO:0071866 [negative regulation of apoptotic process in bone marrow] GO:0071895 [odontoblast differentiation] GO:0071899 [negative regulation of estrogen receptor binding] GO:0090068 [positive regulation of cell cycle process] GO:0090090 [negative regulation of canonical Wnt signaling pathway]
Predicted intracellular proteins Plasma proteins Transcription factors Other all-alpha-helical DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000977 [RNA polymerase II regulatory region sequence-specific DNA binding] GO:0001228 [transcriptional activator activity, RNA polymerase II transcription regulatory region sequence-specific binding] GO:0001569 [patterning of blood vessels] GO:0001649 [osteoblast differentiation] GO:0001755 [neural crest cell migration] GO:0001756 [somitogenesis] GO:0001837 [epithelial to mesenchymal transition] GO:0002040 [sprouting angiogenesis] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003705 [transcription factor activity, RNA polymerase II distal enhancer sequence-specific binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005667 [transcription factor complex] GO:0005737 [cytoplasm] GO:0006366 [transcription from RNA polymerase II promoter] GO:0008013 [beta-catenin binding] GO:0008284 [positive regulation of cell proliferation] GO:0008301 [DNA binding, bending] GO:0010628 [positive regulation of gene expression] GO:0010718 [positive regulation of epithelial to mesenchymal transition] GO:0016055 [Wnt signaling pathway] GO:0016202 [regulation of striated muscle tissue development] GO:0021542 [dentate gyrus development] GO:0021854 [hypothalamus development] GO:0021861 [forebrain radial glial cell differentiation] GO:0021873 [forebrain neuroblast division] GO:0021879 [forebrain neuron differentiation] GO:0021943 [formation of radial glial scaffolds] GO:0022408 [negative regulation of cell-cell adhesion] GO:0022409 [positive regulation of cell-cell adhesion] GO:0030223 [neutrophil differentiation] GO:0030284 [estrogen receptor activity] GO:0030307 [positive regulation of cell growth] GO:0030326 [embryonic limb morphogenesis] GO:0030331 [estrogen receptor binding] GO:0030335 [positive regulation of cell migration] GO:0030509 [BMP signaling pathway] GO:0030854 [positive regulation of granulocyte differentiation] GO:0030879 [mammary gland development] GO:0032696 [negative regulation of interleukin-13 production] GO:0032713 [negative regulation of interleukin-4 production] GO:0032714 [negative regulation of interleukin-5 production] GO:0032993 [protein-DNA complex] GO:0035326 [enhancer binding] GO:0042100 [B cell proliferation] GO:0042393 [histone binding] GO:0043027 [cysteine-type endopeptidase inhibitor activity involved in apoptotic process] GO:0043066 [negative regulation of apoptotic process] GO:0043154 [negative regulation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0043392 [negative regulation of DNA binding] GO:0043401 [steroid hormone mediated signaling pathway] GO:0043565 [sequence-specific DNA binding] GO:0043923 [positive regulation by host of viral transcription] GO:0043966 [histone H3 acetylation] GO:0043967 [histone H4 acetylation] GO:0044212 [transcription regulatory region DNA binding] GO:0045063 [T-helper 1 cell differentiation] GO:0045295 [gamma-catenin binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046632 [alpha-beta T cell differentiation] GO:0048069 [eye pigmentation] GO:0048341 [paraxial mesoderm formation] GO:0048699 [generation of neurons] GO:0048747 [muscle fiber development] GO:0060021 [palate development] GO:0060070 [canonical Wnt signaling pathway] GO:0060325 [face morphogenesis] GO:0060326 [cell chemotaxis] GO:0060561 [apoptotic process involved in morphogenesis] GO:0060710 [chorio-allantoic fusion] GO:0061153 [trachea gland development] GO:0070016 [armadillo repeat domain binding] GO:0070742 [C2H2 zinc finger domain binding] GO:0071345 [cellular response to cytokine stimulus] GO:0071353 [cellular response to interleukin-4] GO:0071864 [positive regulation of cell proliferation in bone marrow] GO:0071866 [negative regulation of apoptotic process in bone marrow] GO:0071895 [odontoblast differentiation] GO:0071899 [negative regulation of estrogen receptor binding] GO:0090068 [positive regulation of cell cycle process] GO:0090090 [negative regulation of canonical Wnt signaling pathway]
Predicted intracellular proteins Plasma proteins Transcription factors Other all-alpha-helical DNA-binding domains Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0000977 [RNA polymerase II regulatory region sequence-specific DNA binding] GO:0001228 [transcriptional activator activity, RNA polymerase II transcription regulatory region sequence-specific binding] GO:0001569 [patterning of blood vessels] GO:0001649 [osteoblast differentiation] GO:0001755 [neural crest cell migration] GO:0001756 [somitogenesis] GO:0001837 [epithelial to mesenchymal transition] GO:0002040 [sprouting angiogenesis] GO:0003677 [DNA binding] GO:0003682 [chromatin binding] GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0003705 [transcription factor activity, RNA polymerase II distal enhancer sequence-specific binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005667 [transcription factor complex] GO:0005737 [cytoplasm] GO:0006366 [transcription from RNA polymerase II promoter] GO:0008013 [beta-catenin binding] GO:0008284 [positive regulation of cell proliferation] GO:0008301 [DNA binding, bending] GO:0010628 [positive regulation of gene expression] GO:0010718 [positive regulation of epithelial to mesenchymal transition] GO:0016055 [Wnt signaling pathway] GO:0016202 [regulation of striated muscle tissue development] GO:0021542 [dentate gyrus development] GO:0021854 [hypothalamus development] GO:0021861 [forebrain radial glial cell differentiation] GO:0021873 [forebrain neuroblast division] GO:0021879 [forebrain neuron differentiation] GO:0021943 [formation of radial glial scaffolds] GO:0022408 [negative regulation of cell-cell adhesion] GO:0022409 [positive regulation of cell-cell adhesion] GO:0030223 [neutrophil differentiation] GO:0030284 [estrogen receptor activity] GO:0030307 [positive regulation of cell growth] GO:0030326 [embryonic limb morphogenesis] GO:0030331 [estrogen receptor binding] GO:0030335 [positive regulation of cell migration] GO:0030509 [BMP signaling pathway] GO:0030854 [positive regulation of granulocyte differentiation] GO:0030879 [mammary gland development] GO:0032696 [negative regulation of interleukin-13 production] GO:0032713 [negative regulation of interleukin-4 production] GO:0032714 [negative regulation of interleukin-5 production] GO:0032993 [protein-DNA complex] GO:0035326 [enhancer binding] GO:0042100 [B cell proliferation] GO:0042393 [histone binding] GO:0043027 [cysteine-type endopeptidase inhibitor activity involved in apoptotic process] GO:0043066 [negative regulation of apoptotic process] GO:0043154 [negative regulation of cysteine-type endopeptidase activity involved in apoptotic process] GO:0043392 [negative regulation of DNA binding] GO:0043401 [steroid hormone mediated signaling pathway] GO:0043565 [sequence-specific DNA binding] GO:0043923 [positive regulation by host of viral transcription] GO:0043966 [histone H3 acetylation] GO:0043967 [histone H4 acetylation] GO:0044212 [transcription regulatory region DNA binding] GO:0045063 [T-helper 1 cell differentiation] GO:0045295 [gamma-catenin binding] GO:0045892 [negative regulation of transcription, DNA-templated] GO:0045893 [positive regulation of transcription, DNA-templated] GO:0045944 [positive regulation of transcription from RNA polymerase II promoter] GO:0046632 [alpha-beta T cell differentiation] GO:0048069 [eye pigmentation] GO:0048341 [paraxial mesoderm formation] GO:0048699 [generation of neurons] GO:0048747 [muscle fiber development] GO:0060021 [palate development] GO:0060070 [canonical Wnt signaling pathway] GO:0060325 [face morphogenesis] GO:0060326 [cell chemotaxis] GO:0060561 [apoptotic process involved in morphogenesis] GO:0060710 [chorio-allantoic fusion] GO:0061153 [trachea gland development] GO:0070016 [armadillo repeat domain binding] GO:0070742 [C2H2 zinc finger domain binding] GO:0071345 [cellular response to cytokine stimulus] GO:0071353 [cellular response to interleukin-4] GO:0071864 [positive regulation of cell proliferation in bone marrow] GO:0071866 [negative regulation of apoptotic process in bone marrow] GO:0071895 [odontoblast differentiation] GO:0071899 [negative regulation of estrogen receptor binding] GO:0090068 [positive regulation of cell cycle process] GO:0090090 [negative regulation of canonical Wnt signaling pathway]