We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
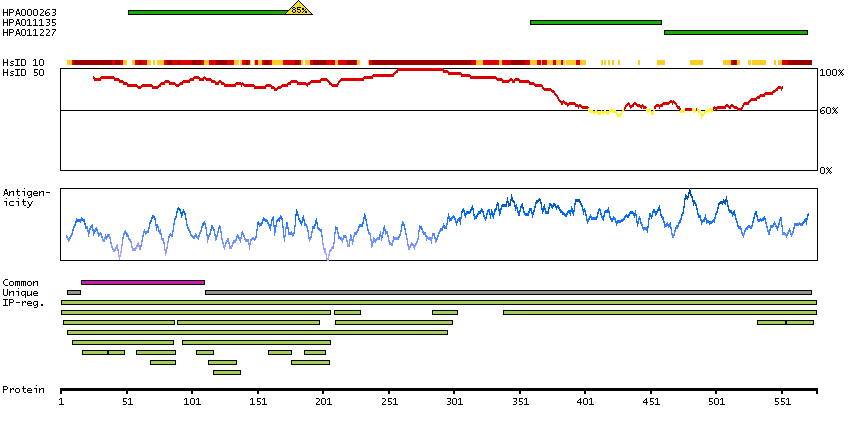
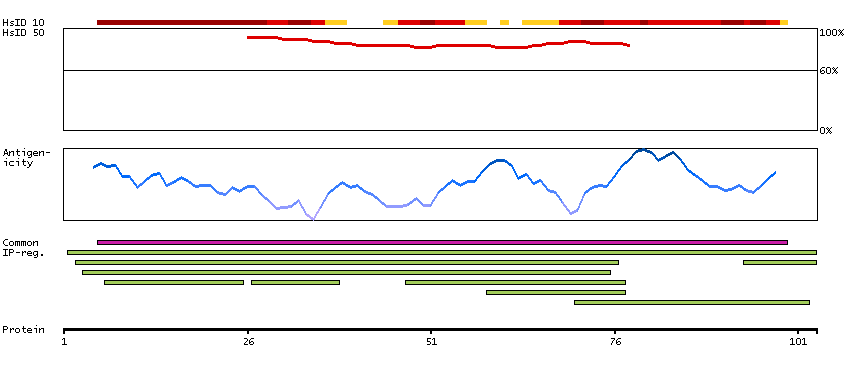
Moesin (for membrane-organizing extension spike protein) is a member of the ERM family which includes ezrin and radixin. ERM proteins appear to function as cross-linkers between plasma membranes and actin-based cytoskeletons. Moesin is localized to filopodia and other membranous protrusions that are important for cell-cell recognition and signaling and for cell movement. [provided by RefSeq, Jul 2008]
Predicted intracellular proteins Plasma proteins Cancer-related genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Predicted intracellular proteins Cancer-related genes Mutational cancer driver genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)