We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
The protein encoded by this gene belongs to the AF4 family of transcription factors involved in leukemia. It is a component of the positive transcription elongation factor b (P-TEFb) complex. A chromosomal translocation involving this gene and MLL gene on chromosome 11 is found in infant acute lymphoblastic leukemia with ins(5;11)(q31;q31q23). [provided by RefSeq, Oct 2011]
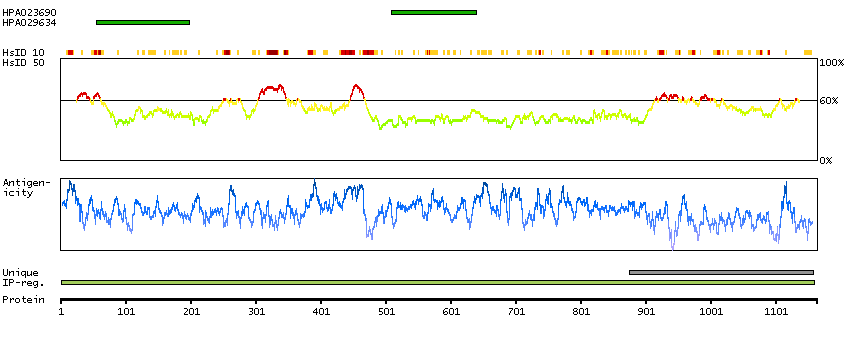
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005739 [mitochondrion] GO:0006355 [regulation of transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0007286 [spermatid development] GO:0008023 [transcription elongation factor complex] GO:0035327 [transcriptionally active chromatin]
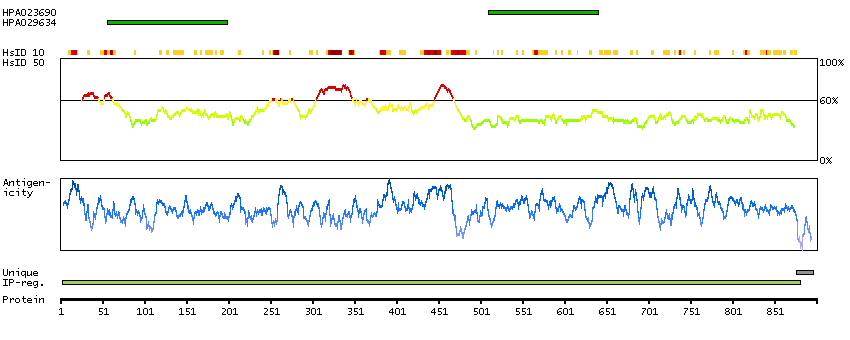
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Disease related genes Protein evidence (Ezkurdia et al 2014)
Show all
GO:0003700 [transcription factor activity, sequence-specific DNA binding] GO:0005515 [protein binding] GO:0005634 [nucleus] GO:0005654 [nucleoplasm] GO:0005730 [nucleolus] GO:0005739 [mitochondrion] GO:0006355 [regulation of transcription, DNA-templated] GO:0006366 [transcription from RNA polymerase II promoter] GO:0008023 [transcription elongation factor complex] GO:0035327 [transcriptionally active chromatin]
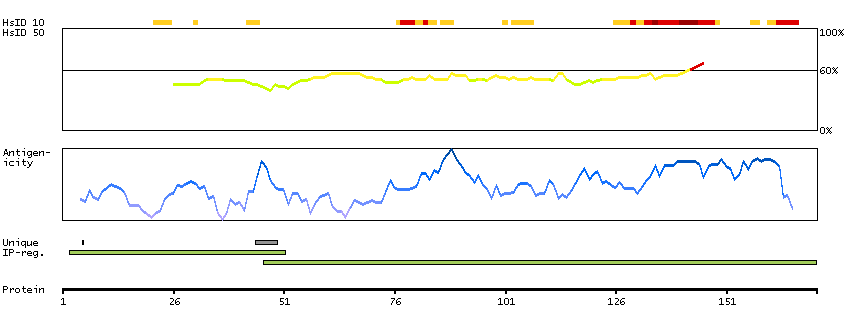
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)
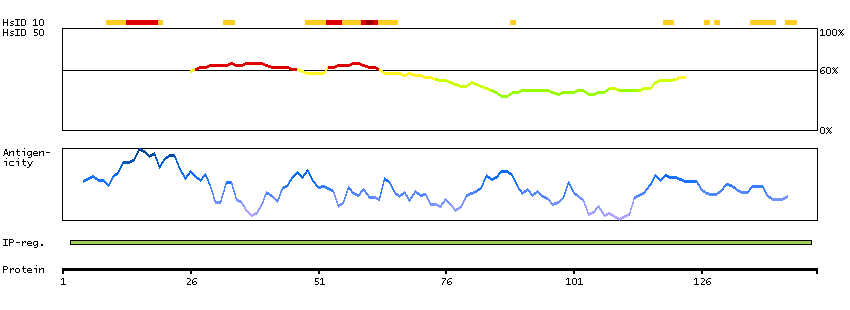
Predicted intracellular proteins Cancer-related genes COSMIC somatic mutations in cancer genes COSMIC Somatic Mutations COSMIC Translocations Protein evidence (Ezkurdia et al 2014)