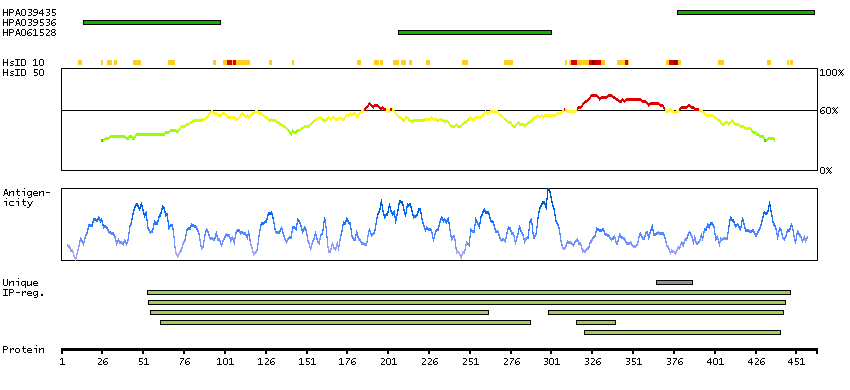
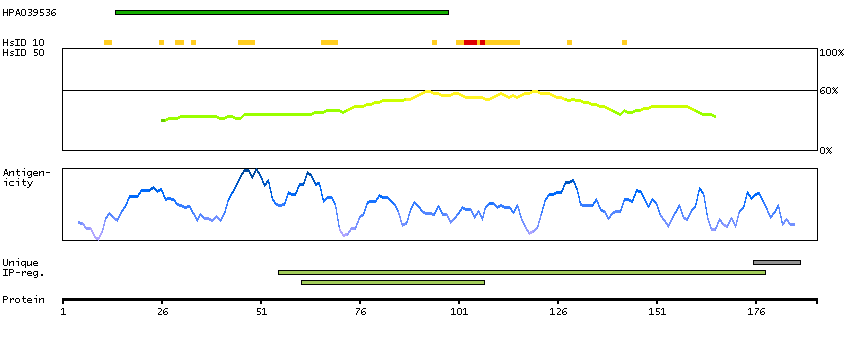
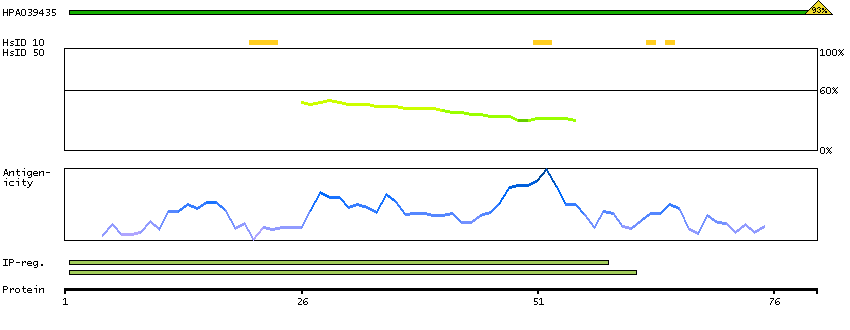
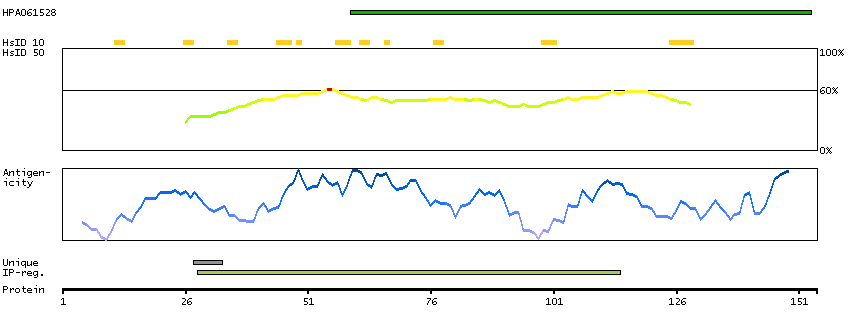
We use cookies to enhance the usability of our website. If you continue, we'll assume that you are happy to receive all cookies. More information. Don't show this again.
Succinyl-CoA synthetase (SCS) is a mitochondrial matrix enzyme that acts as a heterodimer, being composed of an invariant alpha subunit and a substrate-specific beta subunit. The protein encoded by this gene is an ATP-specific SCS beta subunit that dimerizes with the SCS alpha subunit to form SCS-A, an essential component of the tricarboxylic acid cycle. SCS-A hydrolyzes ATP to convert succinate to succinyl-CoA. Defects in this gene are a cause of myopathic mitochondrial DNA depletion syndrome. A pseudogene of this gene has been found on chromosome 6. [provided by RefSeq, Jul 2008]
Enzymes ENZYME proteins Ligase Predicted intracellular proteins Plasma proteins Citric acid cycle related proteins Disease related genes Potential drug targets Protein evidence (Kim et al 2014) Protein evidence (Ezkurdia et al 2014)